

Jest to kontynuacja poszukiwań klasycznych rozwiązań dla wyjaśnienia zjawisk kwantowych i jest to następny krok do mojej poprzedniej notki
http://przestrz.salon24.pl/684602,pierwsze-symulacje-oscylujacych-atomow
Przypomnę że głównym moim założeniem jest występowanie linii pola magnetycznego tuż nad powłoką nukleonów. Jest to w moim pomyśle wytłumaczenie istnienia silnych oddziaływań jądrowych i skoro jest to oddziaływanie ujemne, odpowiada ono również za odpychanie elektronu od jądra na małych odległościach, co jest przyczyną stabilnych powłok elektronowych.
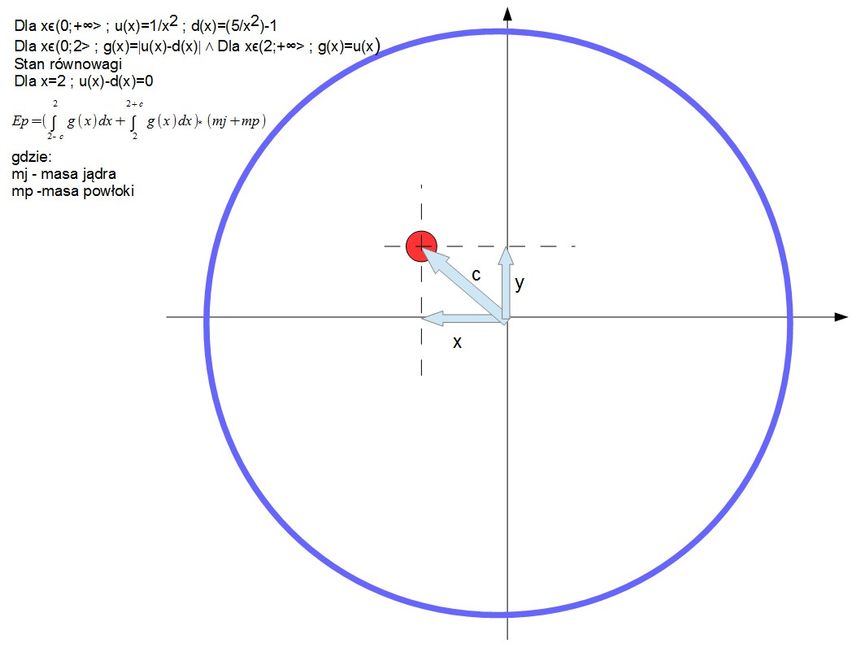
Przedstawione tu modele mają za zadanie w najprostszy sposób przedstawić moje pomysły dlatego też dlatego też stosuje bardzo proste uproszczenia. Proporcje atomu to 1 do 10 masy powłoki do masy jądra i rozmiaru jądra do rozmiaru powłoki. Powłoka jak i jądro jest bryłą sztywną. Układ równowagi uzyskałem poprzez połączenie powłoki z jądrem za pomocą sprężyn. Do pewnych wyliczeń zastosowałem funkcje podobne. Modele te nie odzwierciedlają realnych bytów są jedynie pewnymi uproszczonymi schematami by pokazać pewne zależności.
W pierwszej kolejności wybrałem dwie funkcje wymierne które mają symulować oddziaływania w pobliżu jądra.

Przykładowa funkcja siły oddziaływania ujemnego względem odległości od jądra
Dla xϵ(0;+∞> ; u(x)=1/x2
Przykładowa funkcja siły oddziaływania dodatniego względem odległości od jądra
Dla xϵ(0;+∞> ; d(x)=(5/x2)-1
siła oddziaływań po odjęciu ich wartości
Dla xϵ(0;√5> ; g(x)=|u(x)-d(x)| ˄ Dla xϵ(√5;+∞> ; g(x)=u(x)
Następnym krokiem jest wyliczenie energii potencjalnej w momencie gdy jądro znajduje się poza środkiem równowagi w przestrzeni 2D odseparowanej cząstki. Nie wiem czy nie popełniłem błędu ale udało się to przedstawić jednym wzorem dla jądra i powłoki.

Energię kinetycznej policzyłem już osobno dla jądra i powłoki.


W dalszych obliczeniach przeszedłem już do przestrzeni 1D by nieco uprościć schemat i rozpisałem stan układu dla maksymalnej energii potencjalnej układu względem siebie.

Oraz momenty kiedy powłoka posiada maksymalne i minimalne prędkości właściwe.

Czym jest owa oscylacja w moich pomysłach? Jest to energia wewnętrzna potocznie zwana temperaturą.
Jak widać powłoka może mieć całkiem dużą rozpiętość prędkości właściwej i co za tym idzie jej pędu. Jakie są tego konsekwencje? Przy zderzeniu, cząstka o mniejszej energii całkowitej może mieć większy pęd powłoki od pędu powłoki cząstki z większą energią całkowitą. Istnieje możliwość przekazania energii z cząstki która posiada jej mniej do cząstki która posiada jej więcej. Oczywiście prawdopodobieństwo takiego zdarzenia jest zawsze mniejsze niż odwrotne i zmniejsza się wraz ze zwiększaniem różnicy energii między nimi. W rzeczywistości mamy do czynienia zderzenia elektron, elektron i zgodnie z moimi postulatami w pewnej odległości od siebie zaczynają działać siły odpychające. Samo odbicie również jest ciekawe gdyż nie zachodzi zgodnie z zasadami zderzeń kul a ich kąty odbicia i energie przekazane między sobą będą zależne od fazy oscylacji i będą różne fazy zderzenia, od fazy kumulacji energii potencjalnej po fazę jej oddawania. Bardzo fajnie to widać na symulacjach które przeprowadziłem.
Według mnie sytuacja w której elektrony mają bezpośredni kontakt wymaga bardzo dużych temperatur. Po za tym wszystko powinno być elastyczne, a przedstawione tu modele są sztywne ale nie zmienia to faktu możliwości przekazania energii z cząstki o jej niższym zasobie do cząstki z wyższym.
Przejdźmy teraz do symulacji w programie Algodoo a raczej do błędów tego programu.
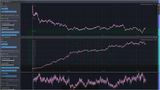
Sprawdziłem energie sumaryczną struktury przy wyłączeniu wszelkich rozproszeń energii. Jak widać na wykresie program przy wyliczeniach zderzeń dodaje nieco energii poza tym nie radzi sobie z jej sumowaniem. Energia sumaryczna cząstki powinna być stała a rośnie i jeszcze oscyluje. Według mojej intuicji program nie umie obliczyć energii oscylacji skręcającej i stąd ta oscylacja. Po jedenastu odbiciach energia układu wzrosła z 140J do 148J.

Pierwszą symulacją z większą ilością atomów jest przejście energii wewnętrznej pomiędzy dwoma komorami gdzie znajduje się po 16 cząstek, przedzielonych dużo cięższymi kulami które są połączone ze sobą i pomieszczeniem sprężynami. Kule symulują przegrodę z materii stałej. Pierwotnie zamiast kul były oscylatory ale program nie radzi sobie i w krótkim czasie dostawały one porządnego kopa energii znikąd. Zastąpiłem je kulami. W pierwszej kolejności sprawdziłem energie sumaryczną wszystkich elementów i tak jak wcześniej mówiłem energia układu miała tendencje wzrostową. Zastosowałem więc współczynnik oporu powietrza tak że po 20 minutach energia układu oscyluje ale nie ma tendencji rosnących ani malejących. Nie jest to najszczęśliwsze rozwiązanie ale na razie nie mam lepszego pomysłu.

Wygenerowałem teraz trzy wykresy:
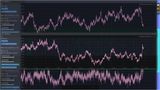
Energia sumaryczna16 cząstek pierwszej komory o większej energii wewnętrznej.
Energia sumaryczna16 cząstek drugiej komory o mniejszej energii wewnętrznej.
Przegroda 12 kul połączonych sprężynami.

Po ponad pięciu minutach energia sumaryczna cząstek w obu komorach uzyskały podobne wartości.
Następnie energia sumaryczna w obydwóch komorach oscylowała wokół pewnej wartości.

Miałem jeszcze opisać generowanie fotonów oraz mój pomysł gdzie znaleźć geometryczną interpretacje serii Lymana. Ale siedzę przy tym cały dzień i mam dość. Proszę o cierpliwość.
Jestem amatorem i całą wiedzę jaką posiadam zdobywam w wolnym czasie dlatego jeżeli widzisz jakiś błąd w rachunkach to będę wdzięczny za jego wskazanie.
Zobacz galerię zdjęć:












Komentarze
Pokaż komentarze (9)