„Pomimo rozległej wiedzy na temat strukturalnych zmian genomicznych towarzyszących ewolucji człowieka nadal nie możemy zidentyfikować genów, które doprowadziły do transformacji małpy w człowieka. Chociaż genetycy opisują geny, które w specyficzny sposób ulegają ekspresji tylko u człowieka nie było i nie ma przełomowych odkryć na polu tych badań.”
 https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-020-06962-8
https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-020-06962-8
 ”Wciąż dużym zainteresowaniem cieszą się cechy genetyczne odróżniające nas ludzi od szympansów i czyniące z nas ludzi (geny odpowiedzialne za to, że jesteśmy ludźmi). Po dywergencji (rozejściu się lini ewolucyjnych od wspólnego przodka człowieka i szympansa) genom człowieka i szympansa przeszedł wiele zmian w postaci: substytucji (mutacji punktowych) pojedynczych nukleotydów, delecji (ubytków DNA), duplikacji fragmentów DNA (mutacja zwana duplikacją tworzy kopię jakiegoś fragmentu DNA - na przykład genu - na tym samym chomosomie) o różnej długości. Insercji (wstawień fragmentów łańcucha DNA) ruchomych elementów genetycznych (transpozonów) z jednego miejsca genomu do innego (na zasadzie kopiuj-wklej) oraz rearanżacji chromosomów.
”Wciąż dużym zainteresowaniem cieszą się cechy genetyczne odróżniające nas ludzi od szympansów i czyniące z nas ludzi (geny odpowiedzialne za to, że jesteśmy ludźmi). Po dywergencji (rozejściu się lini ewolucyjnych od wspólnego przodka człowieka i szympansa) genom człowieka i szympansa przeszedł wiele zmian w postaci: substytucji (mutacji punktowych) pojedynczych nukleotydów, delecji (ubytków DNA), duplikacji fragmentów DNA (mutacja zwana duplikacją tworzy kopię jakiegoś fragmentu DNA - na przykład genu - na tym samym chomosomie) o różnej długości. Insercji (wstawień fragmentów łańcucha DNA) ruchomych elementów genetycznych (transpozonów) z jednego miejsca genomu do innego (na zasadzie kopiuj-wklej) oraz rearanżacji chromosomów.
Specyficzne dla człowieka, odróżniające genom człowieka od genomu szympansa, zmiany w ludzkim DNA, polegające na zamianie jednego nukleotydu na inny (mutacja punktowa) stanowią 1,23% ludzkiego DNA (genomu), podczas gdy delecje i insercje obejmują ~ 3% naszego genomu. Co więcej, znacznie większy odsetek zmian właściwych ludziom stanowią zróżnicowane inwersje (mutacje polegające na odwróceniu jakiegoś fragmentu łańcucha DNA) i translokacje chromosomów (translokacja polega na oderwaniu się dużego fragmentu chromosomu i przyczepieniu się w inne miejsce tego chromosomu, lub do innego chromosomu) obejmujące kilka regionów o długości megazasad lub nawet całych chromosomów!
Jednak pomimo rozległej wiedzy na temat strukturalnych zmian genomicznych towarzyszących ewolucji człowieka nadal nie możemy zidentyfikować genów, które doprowadziły do transformacji małpy w człowieka. Chociaż genetycy opisują geny, które w specyficzny sposób ulegają ekspresji tylko u człowieka nie było i nie ma przełomowych odkryć na polu tych badań.
Większość zmian strukturalnych, które doprowadziły do transformacji małpy w człowieka musiała zajść na poziomie wyższym poziomie regulatorów ekspresji genów, co z kolei musiało doprowadzić do zmian w sieciach genetycznych oraz w integracjach między sieciami genetycznymi (w oddziaływaniach epistatycznych podczas wysokiego poziomu epistazy). W tym przeglądzie podsumowaliśmy dostępne informacje na temat różnic genetycznych między ludźmi i szympansami oraz ich potencjalnego wpływu funkcjonalnego na zróżnicowane cechy molekularne, anatomiczne, fizjologiczne i psychiczne (behawioralne) tych gatunków.
(.....)
Obecnie powszechnie przyjmuje się, że nie tylko zmiany na poziomie genów kodujących białka (gdzie znaleziono wiele podobieństw między ludźmi i szympansami), ale przede wszystkim zmiany w sekwencjach genetycznych odpowiedzialnych za regulację ich ekspresji odegrały główną rolę w kształtowaniu się różnic fenotypowych między ludźmi i szympansami (.....)”
 Geny kodujące białka, których człowiek posiada 30 000 z czego 20 do 25 000 koduje białka, to jedynie cegiełki. Natomiast geny regulujące ich ekspresję stanowią przepis w jaki sposób zbudować z nich: kleszcza, mysz, psa, szympansa czy człowieka. W tych genach należy szukać przepisu na człowieka.
Geny kodujące białka, których człowiek posiada 30 000 z czego 20 do 25 000 koduje białka, to jedynie cegiełki. Natomiast geny regulujące ich ekspresję stanowią przepis w jaki sposób zbudować z nich: kleszcza, mysz, psa, szympansa czy człowieka. W tych genach należy szukać przepisu na człowieka.
Uczeni poznali i zrozumieli około 2% ludzkiego DNA. Naukowcy nadal nie wiedzą co dokładnie znajduje się w pozostałych 98% DNA człowieka. Większość ludzkiego DNA nie koduje białek. Pośród tego niekodującego DNA ukrytych jest przed oczami naukowców wiele kluczowych elementów regulacyjnych, które kontrolują aktywność tysięcy genów, które ulegają ekspresji na różne sposoby w rozmaitych komórkach budujących ludzkie ciało.
Szacuje się, że jest ich razem około 10 bilionów! Inne poznane geny kodują różne formy RNA: rybosomowe RNA, tRNA, które zawierają antykodony indentyfikujące kodony zawarte w matrycowym RNA podczas translacji. Do różnych cząsteczek tRNA jest przypisywany konkretny aminokwas następnie jest on przyłączany do wydłużającego się polipeptydu w procesie syntezy białka (podczas translacji).
Inny rodzaj zakodowanych cząsteczek RNA, to regulatorowe cząsteczki RNA. Regulują ekspresję genów na poziomie translacji.
Potrzebna jest do tego maszyneria zbudowana z białek zakodowana pośród zbadanych 300 000 genów. Chociaż ludzki genom zawiera około 30 000 genów, to istnieje prawdopodobnie od 500 000 do 1 000 000 rozmaitych rodzajów ludzkich białek!
Średnio na gen przypada 40 białek, jeśli człowiek wytwarza 1 milion białek i ma tylko 25 000 genów. Jeśli człowiek wytwarza 1 milion białek, ale ludzkie DNA koduje tylko 25 000 genów, możemy obliczyć średnią liczbę białek wytwarzanych na gen, po prostu dzieląc całkowitą liczbę białek przez liczbę genów. Jest to proste obliczenie matematyczne stosowane do zrozumienia związku między genami i białkami w biologii człowieka. Aby więc znaleźć średnią liczbę białek wytwarzanych przez jeden gen podziel 1 000 000 białek przez 25 000 genów. Średnia liczba białek na gen = 40 białek na gen. Dlaczego w 25 000 genów można zakodować tyle białek? Jak to możliwe?
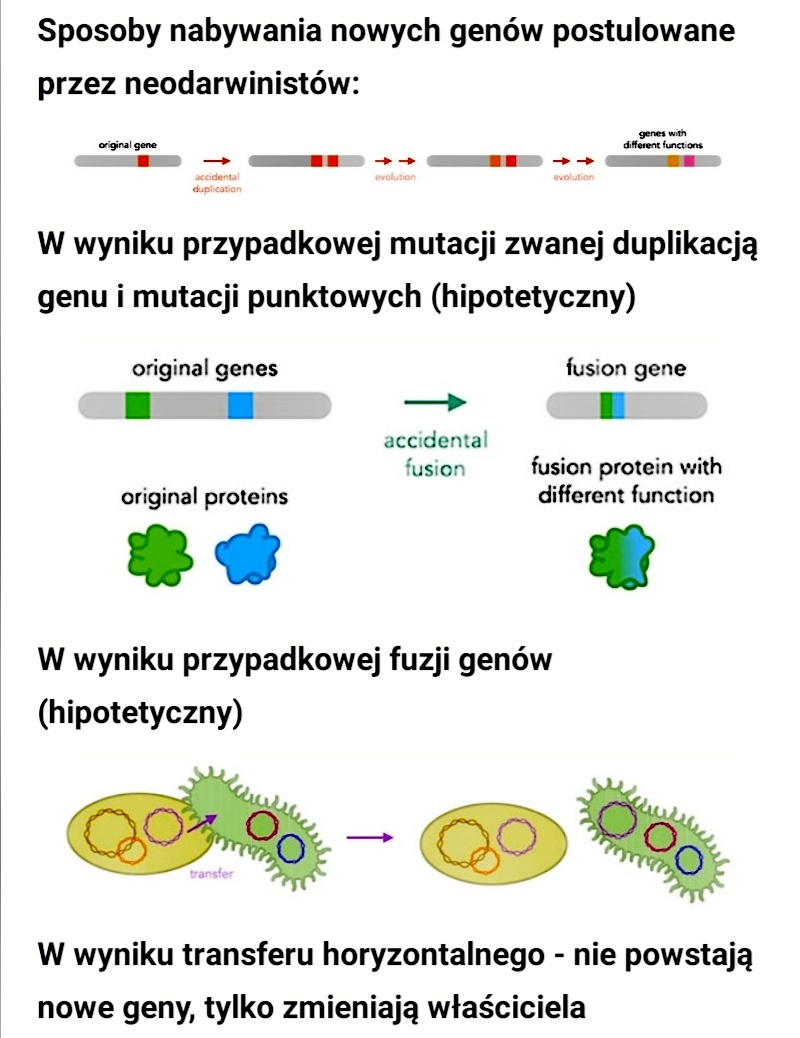
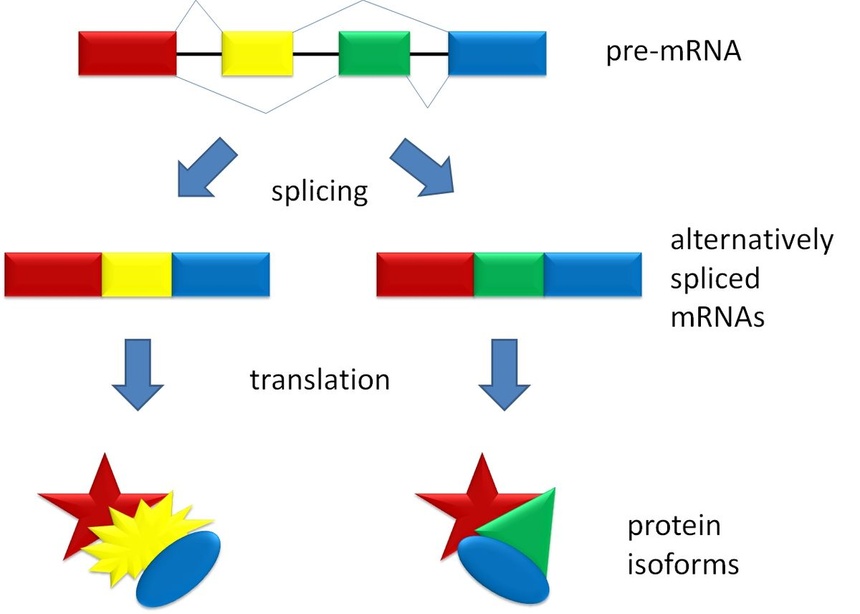
Dawniej popularne było twierdzenie: jeden gen - jedno białko. Obecnie wiadomo, że na matrycy jednego genu w DNA może powstać więcej niż jedno białko. Naukowcy zidentyfikowali wiele mechanizmów, ktore są za to odpowiedzialne: alternatywne składanie, nakładające się geny czy różnego rodzaju rekombinacje.
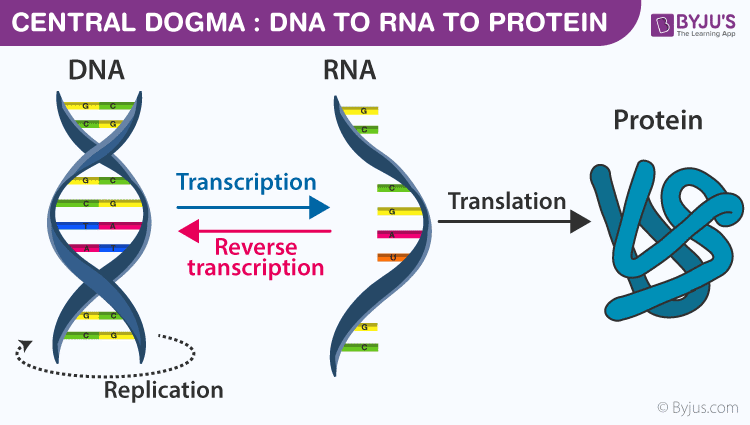
https://byjus.com/biology/central-dogma-inheritance-mechanism/
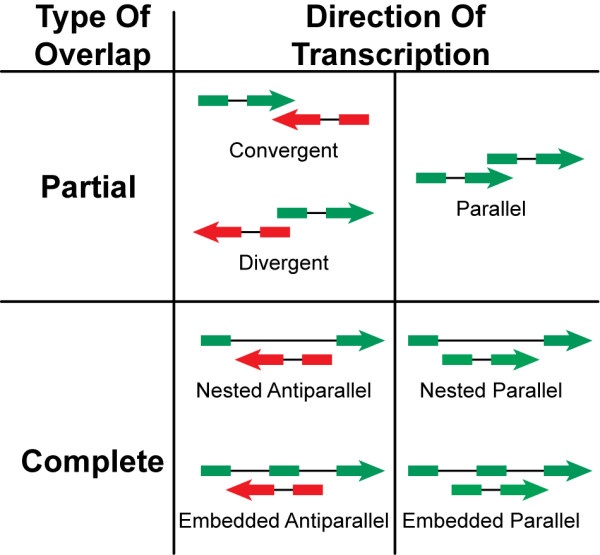
https://www.researchgate.net/figure/Classes-of-overlapping-genes-OGC-classification-was-based-on-the-overlap-extent_fig1_5435467
 https://www.nature.com/scitable/topicpage/eukaryotic-genome-complexity-437/
https://www.nature.com/scitable/topicpage/eukaryotic-genome-complexity-437/
„Poza szacowaniem liczby genów kodujących białka Podobnie jak w przypadku wielkości genomu, posiadanie większej liczby genów kodujących białka niekoniecznie przekłada się na większą złożoność. Dzieje się tak, ponieważ genom eukariotyczny ewoluował w inny sposób, aby generować złożoność biologiczną. Duża część tej złożoności wynika z „zachowania” genomu, a dokładniej, z ekspresji różnych genów. Alternatywne składanie było pierwszym zjawiskiem odkrytym przez naukowców, które uświadomiło im, że złożoności genomu nie można ocenić na podstawie liczby genów kodujących białka. Podczas alternatywnego splicingu, który ma miejsce po transkrypcji i przed translacją, introny są usuwane, a eksony są łączone ze sobą, tworząc cząsteczkę mRNA. Jednak nie wszystkie eksony muszą być ponownie złożone w ten sam sposób. Zatem pojedynczy gen, czyli jednostka transkrypcyjna, może kodować wiele białek lub innych produktów genów, w zależności od sposobu ponownego złożenia eksonów. W rzeczywistości naukowcy oszacowali, że może istnieć aż 500 000 lub więcej różnych ludzkich białek, a wszystkie kodowane są przez zaledwie 20 000 genów kodujących białka”.
https://simple.m.wikipedia.org/wiki/Alternative_splicing
 Chociaż dużo zostało jeszcze do odkrycia, to już teraz wiadomo gdzie szukać genów regulatorowych, które umożliwiają syntezę setek tysięcy białek strukturalnych i odpowiedzialnych za sygnalizację komórkową, która reguluje ekspresję genów.
Chociaż dużo zostało jeszcze do odkrycia, to już teraz wiadomo gdzie szukać genów regulatorowych, które umożliwiają syntezę setek tysięcy białek strukturalnych i odpowiedzialnych za sygnalizację komórkową, która reguluje ekspresję genów.
Regulacja ekspresji genów odbywa się na poziomie transkrypcji, translacji czy na poziomie epigenetycznym. Wiadomo też, że przepis na wyrażanie na różne sposoby genów, które znajdują się w 2% ludzkiego genomu znajduje się w tych 98% genomu, który naukowcy dopiero zaczynają poznawać. Przypomnę, że największych podobieństw między ludźmi i szympansami dopatrzono się w tych 2% ludzkiego genomu i na tej podstawie się twierdzi, że DNA ludzi i szympansów łączy 90,9 % podobieństw. Kolosalne różnice fenotypowe różniące ludzi i szympansy zawiera pozostałe 98% niezbadanego genomu a nie 2% poznanego i porównanego!
https://zpe.gov.pl/a/pochodzenie-i-ewolucja-czlowieka/D14dDU6Qo
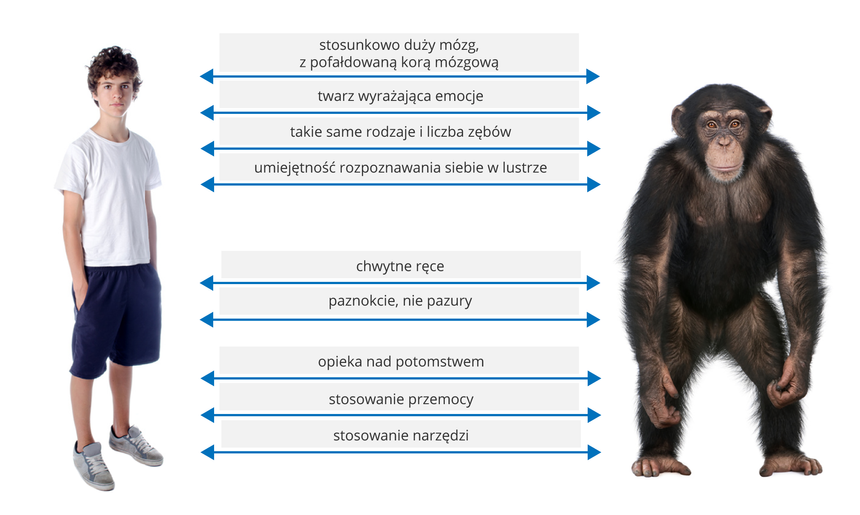
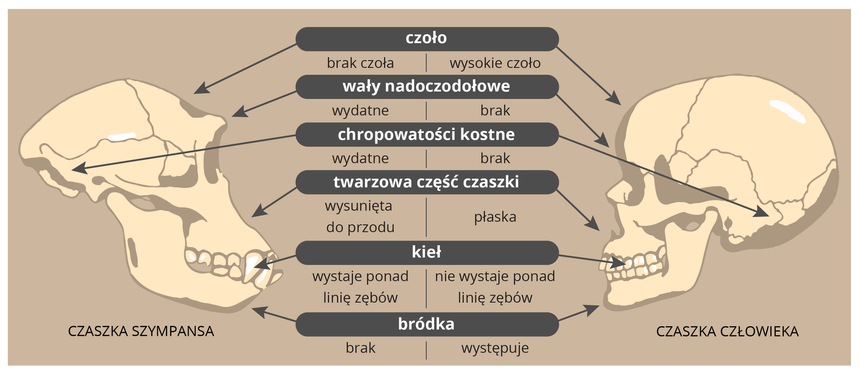 Podobieństwa zaobserwowane Podczas tych porównań w około 30 000 genów ludzi i szympansów nie musiały powstać losowo i zostać odziedziczonymi po wspólnym przodku! Nie bierze się pod uwagę nielosowych, powtarzalnych mutacji w gorących miejscach, które mogą zmylić filogenetyków molekularnych co, do pokrewieństw filogenetycznych oraz zafałszować wnioski, które płyną z koncepcji zegara molekularnego. Szeroko rozpisałem się na ten temat tu: Adres do oryginalnej pracy naukowej z jakiej korzystałem:
Podobieństwa zaobserwowane Podczas tych porównań w około 30 000 genów ludzi i szympansów nie musiały powstać losowo i zostać odziedziczonymi po wspólnym przodku! Nie bierze się pod uwagę nielosowych, powtarzalnych mutacji w gorących miejscach, które mogą zmylić filogenetyków molekularnych co, do pokrewieństw filogenetycznych oraz zafałszować wnioski, które płyną z koncepcji zegara molekularnego. Szeroko rozpisałem się na ten temat tu: Adres do oryginalnej pracy naukowej z jakiej korzystałem:
https://frontlinegenomics.com/genetic-mutations-may-not-be-random/
„Wyniki nowego badania radykalnie podważają neodarwinowską teorię ewolucji. Przez lata wierzono, że mutacje genetyczne zachodzą losowo. Według nowych badań mogą być znacznie mniej przypadkowe, niż początkowo sądzono.
(Syntetyczna) Teoria ewolucji
Powszechnie przyjmuje się pogląd, że mutacje zachodzą w genomie losowo. W rzeczywistości obecna teoria ewolucji opiera się na tej przypadkowości (przypadkowej mutagenezie i losowych czynnikach selekcyjnych). Neodarwiniści twierdzą, że dobór naturalny działa na przypadkową zmienność genetyczną, w wyniku czego pewne cechy stają się mniej lub bardziej powszechne w populacji.
Jednak ostatnie badania wykazały, że wiele czynników takich jak skład nukleotydów może wpływać na prawdopodobieństwo wystąpienia mutacji w określonych lokalizacjach genomu. Poprzednie badania dostarczyły dowodów na to, że naprawa DNA jest ukierunkowana na określone regiony genomu. Zespół odpowiedzialny za nowe badanie dokładniej zbadał te koncepcje, aby dowiedzieć się, czy mutacje genetyczne są rzeczywiście przypadkowe.
Nielosowe współczynniki mutacji
Naukowcy pobrali DNA z roślin Arabidopsis thaliana - znanych również jako rzeżucha talarska. Następnie ustalili duże zestawy mutacji powstałe de novo u rzeżuchy zwyczajnej. Dokonano tego, aby upewnić się, że wszelkie znalezione mutacje nie zostały jeszcze poddane selekcji naturalnej. (....)”
Tu jest reszta informachi na ten temat:
https://www.salon24.pl/u/slawekp7/1361321,szlak-syntezy-witaminy-c-pseudogeny-i-zagadkowy-gen-glo
 „ (....) W tym kontekście złożone podejścia bioinformatyczne łączące różne analizy danych stają się kluczem do znalezienia elementów genetycznych, które przyczyniły się do ewolucji człowieka z małpy. Niezwykle ważne jest również posiadanie odpowiednich modeli eksperymentalnych w celu walidacji potencjalnych zmian genomicznych specyficznych dla gatunku ludzkiego i szympansa.
„ (....) W tym kontekście złożone podejścia bioinformatyczne łączące różne analizy danych stają się kluczem do znalezienia elementów genetycznych, które przyczyniły się do ewolucji człowieka z małpy. Niezwykle ważne jest również posiadanie odpowiednich modeli eksperymentalnych w celu walidacji potencjalnych zmian genomicznych specyficznych dla gatunku ludzkiego i szympansa.
(.....)”
Walidacja – w naukach technicznych i informatyce działanie mające na celu potwierdzenie w sposób udokumentowany i zgodny z założeniami, że procedury, procesy, urządzenia, materiały, czynności i systemy rzeczywiście prowadzą do zaplanowanych wyników.-za wikipedią
 „(....) Obecnie rozwijające się metody eksperymentalne, takie jak uzyskiwanie pluripotencjalnych komórek macierzystych i docelowe modyfikacje genomu, takie jak CRISPR-CAS otwierają ekscytujące perspektywy, które być może pozwolą uczonym na znalezienie przysłowiowej „igły w stogu siana”, która była naprawdę istotna dla ewolucji funkcjonalnej człowieka z małpiego przodka. W rzeczywistości prawdopodobnie przed naukowcami stoi zadanie odszukanie wielu takich „igieł w stogu siana”!
„(....) Obecnie rozwijające się metody eksperymentalne, takie jak uzyskiwanie pluripotencjalnych komórek macierzystych i docelowe modyfikacje genomu, takie jak CRISPR-CAS otwierają ekscytujące perspektywy, które być może pozwolą uczonym na znalezienie przysłowiowej „igły w stogu siana”, która była naprawdę istotna dla ewolucji funkcjonalnej człowieka z małpiego przodka. W rzeczywistości prawdopodobnie przed naukowcami stoi zadanie odszukanie wielu takich „igieł w stogu siana”!
Jednakże, przynajmniej na razie, stosowanie tych eksperymentalnych podejść w odniesieniu do milionów omawianych tutaj potencjalnie istotnych cech specyficznych dla gatunku człowieka i szympansa jest niemożliwe ze względu na wysokie koszty i potrzebe wykonania ogromnej pracy.
Z kolei alternatywnym podejściem mogłoby być połączenie dopracowanych danych w realistyczny model - projekt badawczy przy użyciu podczas badań biologii systemowej nowej generacji. Model ten powinien zostać wypracowany na dużych zbiorach danych genomowych ludzi i innych naczelnych.
Takie podejście mogłoby zintegrować wiedzę na temat interakcji białko-białko, szlaków biochemicznych (sygnalizacyjnych i odpowiedzialnych za metabolizm), czasoprzestrzennych wzorców epigenetycznych, transkryptomicznych i proteomicznych, a także wysokowydajną symulację zmian funkcjonalnych spowodowanych zmienionymi strukturami białek.
https://pin.it/16x8DhV8e
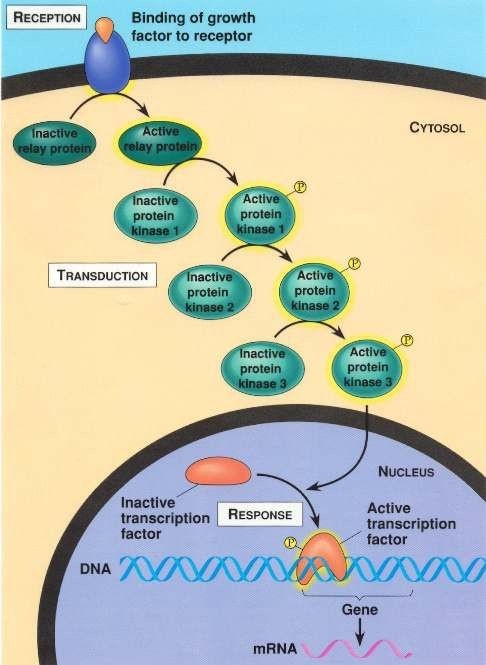 Ujawnione różnice można również analizować w kontekście trendów ewolucyjnych specyficznych dla ssaków i innych naczelnych np. poprzez zastosowanie odpowiedniego podejścia do pomiaru tempa ewolucji zmian strukturalnych w białkach oraz wzbogacenia o elementy transpozycyjne w funkcjonalnych loci genomowych w celu oszacowania ewolucji regulacyjnej genów.
Ujawnione różnice można również analizować w kontekście trendów ewolucyjnych specyficznych dla ssaków i innych naczelnych np. poprzez zastosowanie odpowiedniego podejścia do pomiaru tempa ewolucji zmian strukturalnych w białkach oraz wzbogacenia o elementy transpozycyjne w funkcjonalnych loci genomowych w celu oszacowania ewolucji regulacyjnej genów.
Oprócz analizy danych na poziomie pojedynczego genu informacje te można agregować, aby przyjrzeć się całym procesom organizmowym, rozwojowym lub wewnątrzkomórkowym, np. za pomocą analizy wzbogacania terminów dotyczących ontologii genetycznych i analizy ilościowej szlaków (.....).”
Mit „śmieciowego DNA” – Transpozony pełnią ważne funkcje w genomach zwierząt i roślin podczas regulacji ekspresji genów. Ich działanie regulowane jest przez mechanizmy epigenetyczne!
https://www.ncn.gov.pl/sites/default/files/listy-rankingowe/2018-09-14/streszczenia/428977-pl.pdf


https://www.tandfonline.com/doi/full/10.1080/07352689.2021.1920731
„Przeplatane powtórzenia zwane elementami transpozycyjnymi (TE), powszechnie określanymi jako elementy ruchome, stanowią znaczną część genomów zwierząt wyższych. TE przyczyniają się do kontrolowania ekspresji genów lokalnie, a nawet daleko, na poziomie transkrypcyjnym i potranskrypcyjnym, co jest jednym z ich znaczących efektów funkcjonalnych wpływających na funkcję genów i ewolucję genomu (słowo ewolucja ma w biologii różne znaczenia. Postuluje się ewolucję losową neodarwinowską. Do tej pory wykryto tylko nielosową: naturalną biotechnologię komórkową, w którą zaangażowanych jest mnóstwo wyrafinowanych maszyn molekularnych). Istnieją różne mechanizmy, dzięki którym TE kontrolują ekspresję genów.
Po pierwsze, TE oferują regiony regulatorowe cis w genomie z ich nieodłącznymi cechami regulacyjnymi dla własnej ekspresji, co czyni je potencjalnymi czynnikami kontrolującymi ekspresję genów gospodarza. Elementy promotora i wzmacniacza zawierają miejsca regulatorowe cis wygenerowane z TE, które działają jako miejsca wiązania dla różnych czynników działających w pozycji trans.
Po drugie, wykazano, że znaczna część miRNA i długich niekodujących RNA (lncRNA) ma TE, które kodują regulatorowe RNA, co ujawnia pochodzenie TE tych RNA. Ponadto wykazano, że sekwencje TE są niezbędne dla działań regulacyjnych tych RNA, które obejmują wiązanie z docelowym mRNA. Będąc członkiem cis-regulacyjnych i regulatorowych sekwencji RNA, TE odgrywają zatem istotne role regulacyjne. Ponadto zasugerowano, że pochodzące z TE regulatorowe RNA i regiony cis-regulacyjne przyczyniają się do ewolucyjnej nowości w regulacji genów (nie jest to losowa ewolucja neodarwinowska, tylko nielosowa w ramach normy reakcji na środowisko).”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3562835/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3562835/
„Elementy transpozycyjne (TE) są wszechobecne w genomach roślin i często stanowią znaczną część jądrowego DNA. Na przykład około 40% genomu ryżu składa się z TE, z których wiele to retrotranspozony, w tym 14% retrotranspozonów LTR i ~1% innych niż LTR. Pomimo ich szerokiego rozmieszczenia i liczebności, stwierdzono, że bardzo niewiele TE ma charakter transpozycyjny, co wskazuje, że aktywność TE może być ściśle kontrolowana przez genom gospodarza, aby zminimalizować potencjalnie mutagenne skutki związane z aktywną transpozycją.
Zgodnie z tym poglądem coraz więcej dowodów sugeruje, że epigenetyczne szlaki wyciszania, takie jak metylacja DNA, interferencja RNA i H3K9me2, działają wspólnie w celu tłumienia aktywności TE na poziomach transkrypcyjnych i potranskrypcyjnych. Nie jest jednak jeszcze jasne, czy usunięcie modyfikacji histonów związanych z aktywną transkrypcją jest również zaangażowane w wyciszanie TE. (....)”
https://pubmed.ncbi.nlm.nih.gov/2825167/
„Transpozon Tn10 wstawia preferencyjnie w określonych „gorących punktach” insercji, które mają wspólną symetryczną sekwencję konsensusową 6 par zasad: 5' GCTNAGC 3'. Białko rozpoznające tę sekwencję nie jest znane, ale prawdopodobnie jest to białko transpozazy kodowane przez Tn10. Przedstawiamy dowody na to, że grupy 5-metylowe dwóch tymin w tej sekwencji są niezbędne do skutecznej insercji transpozonu; w przypadku ich braku sekwencja jest nadal rozpoznawana, ale z mniejszą wydajnością. Doszliśmy do tego wniosku badając konkretny gorący punkt, którego sekwencja to 5' GCCAGGC 3'. Tak się składa, że najbardziej wewnętrzne cytozyny tej sekwencji są substratami do metylacji w ich 5 pozycjach przez bakteryjną metylazę kodowaną przez dcm. Stwierdziliśmy, że Tn10 przenosi się do tego miejsca 15 razy częściej (....)”
https://slawekp7.wordpress.com/2021/02/23/2481/
Biolodzy przez dłuższy czas utrzymywali, że DNA zawiera tylko i wyłącznie instrukcje dotyczące syntezy białek. Okazało się jednak, że fragmenty kodujące białka stanowią zaledwie około 2% genomu. Po co nam pozostałe 98% DNA? Jak powiedział John Mattick z Instytutu Biologii Molekularnej Uniwersytetu Stanu Queensland w Brisbane (Australia), tę zagadkową część DNA „natychmiast zaczęto uważać za ewolucyjne śmieci”. Co nikogo nie powinno dziwić, ponieważ z powodu chronicznego braku postępów w biologii ewolucyjnej, jej zwolennicy są zmuszeni do szukania „dowodów” na ewolucję w lukach ludzkiej wiedzy. A więc neodarwiności wymyślili sobie, że rozwojem złożonych organizmów zarządza jedynie jakieś 2% DNA, natomiast 98% to śmieci, które ewentualnie mogą być wykorzystywane, jako materiał, na którym pracuje dobór naturalny. Spekulacjom nie było końca. Np. ruchome elementy DNA, tak zwane transpozony czy retrotranspozony, traktowano jak pasożytnicze DNA, które porównywano np. do tasiemców. Teraz wiadomo, że biorą one udział w regulacji ekspresji genów. Jak się okazało mechanizmy epigenetyczne, a najprawdopodobniej wiele jeszcze nieodkrytych, które z tymi mechanizmami współpracują, są odpowiedzialne za takie procesy, jak różnicowanie się pod względem dostosowania i morfologii, specjacji czy za odzyskiwanie wzroku przez ryby jaskiniowe, których potomstwo powróciło do oświetlonego środowiska.
Jak ta koncepcja wpłynęła na badania genetyków? Zdaniem biologa molekularnego Wojciecha Makałowskiego wspomniany pogląd „odstręczył większość badaczy od zajmowania się niekodującym („śmieciowym”) DNA”, a więc neodarwiniści wyraźnie przyczyniali się do braku w postępie naukowym. Tylko nieliczni naukowcy,narażając się na wyśmianie, zgłębiali to niepopularne zagadnienie. Dzięki ich wysiłkom na początku lat dziewięćdziesiątych XX wieku zaczęto inaczej postrzegać „śmieciowe” DNA”. Makałowski podkreśla, że to, co niegdyś uważano za „śmieci”, teraz biolodzy zazwyczaj nazywają „genomowymi klejnotami”.
Jak twierdzi John Mattick, teoria o „śmieciowym” DNA to klasyczny przykład „ignorowania faktów przez dominujący trend w nauce”. Dodaje też: „Niezauważone następstwa tych faktów (…) [mogą] z powodzeniem przejść do historii jako jedna z największych pomyłek w historii biologii molekularnej” (Świat Nauki, grudzień 2003). Nie ulega zatem wątpliwości, że prawdę naukową trzeba ustalać na podstawie dowodów, a nie — popularnych poglądów.
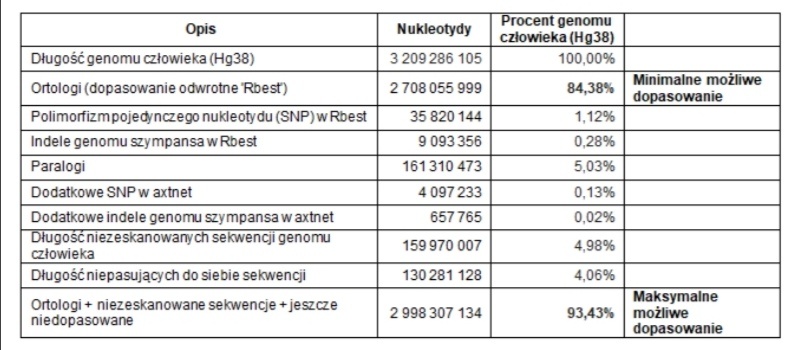
 „(....) I wreszcie, większość opisanych tutaj wyników uzyskano dla genomów referencyjnych człowieka i szympansa, z których każdy został zbudowany przy użyciu DNA kilku osobników. Obecnie większa dostępność sekwencjonowania całego genomu uwypukliła kolejne wyzwanie w porównywaniu ludzi i szympansów – różnorodność genomu populacyjnego.
„(....) I wreszcie, większość opisanych tutaj wyników uzyskano dla genomów referencyjnych człowieka i szympansa, z których każdy został zbudowany przy użyciu DNA kilku osobników. Obecnie większa dostępność sekwencjonowania całego genomu uwypukliła kolejne wyzwanie w porównywaniu ludzi i szympansów – różnorodność genomu populacyjnego.
Na przykład ostatnie badanie obejmujące 910 rodzimych genomów ludzi z Afryki skupiało się na frakcji sekwencji nieobecnych w referencyjnym zestawie genomu Hg38. Zidentyfikowano aż 125 715 brakujących wstawek w Hg38, przy średniej liczbie 859 wstawek na osobę, co daje łącznie 296,5 Mb. Odkrycia te wyraźnie sugerują, że w obecnej wersji złożenia ludzkiego genomu może brakować prawie 10% informacji o ludzkim genomie!
Co więcej, odzwierciedla to również wysoki stopień heterogeniczności genomu populacji afrykańskiej. Podobne badania przeprowadzono także dla innych populacji. Na przykład w populacji chińskiej odkryto łącznie 29,5 Mb nowego DNA i 167 przewidywanych nowych genów, których brakowało w zestawie genomu referencyjnego
Szympansy wykazują również znaczną różnorodność genomu z wieloma cechami specyficznymi dla populacji: szympansy z centralnej Afryki zachowują największą różnorodność w linii szympansów, podczas gdy inne podgatunki wykazują liczne oznaki wąskich gardeł populacyjnych.
Dotychczas nie opublikowano zbyt wielu badań na temat niereferencyjnego porównania genomu człowieka i szympansa. Można jednak dokonać pewnych szacunków. W niedawnym badaniu 1000 genomów ludzi z populacji szwedzkiej zidentyfikowano łącznie 61 044 skupisk tworzących w sumie ~ 46 Mb ludzkiego DNA, których nie było w referencyjnym zespole ludzkiego genomu Hg38. Klastry te autorzy nazwali „nowymi sekwencjami” (NS). Zgodnie z oczekiwaniami NS zostały wzbogacone o proste powtórzenia i satelity i znacznie różniły się u poszczególnych osób.
Większą część NS (32 794) dopasowano do sekwencji niereferencyjnych z wyżej wymienionego badania 910 afrykańskich genomów. Ostatecznie aż 18 773 NS było obecnych także w zespole genomu PT4 szympansa. Jeśli chodzi o sekwencje kodujące białka, to 143 ortologiczne geny szympansów zawierały łącznie 2807 NS przy czym silnie wzbogacone były cztery geny: EPPK1, OR8U1, NINL i METTL21C. Pozycjonowanie insercji NS w ludzkim genomie ujawniło, że 2195 z nich zlokalizowano w obrębie 2384 genów, przy czym 85 insercji NS znaleziono w eksonach 82 genów.
Inne konsorcjum badawcze badało niepowtarzające się sekwencje niereferencyjne (NRNR) w genomach 15 219 Islandczyków. Znaleziono łącznie 326 596 par zasad DNA NRNR, z czego ~ 84% powstało jedynie przez 244 insercje dłuższe niż 200 bp. Warto zauważyć, że porównanie z genomem szympansa ujawniło, że ponad 95% NRNR dłuższych niż 200 bp było również obecnych w zespole genomu szympansa, co wskazuje, że byli to przodkowie. Zatem brak informacji na temat różnorodności populacji genomu może mieć wpływ na całkowity zakres rozbieżności międzygatunkowych ludzi i szympansów poprzez błędną interpretację sekwencji polimorficznych (....)”
Najczęstszą formą zmienności genetycz- nej jest polimorfizm pojedynczego nukleotydu (SNP, single nucleotide polymophism)
 „(.....) Nie unieważnia to jednak większości hipotez i faktów wspomnianych w tej recenzji. Mimo to odkrycia te nieuchronnie prowadzą do idei potrzeby i porównania pangenomów człowieka iszympansa.”
„(.....) Nie unieważnia to jednak większości hipotez i faktów wspomnianych w tej recenzji. Mimo to odkrycia te nieuchronnie prowadzą do idei potrzeby i porównania pangenomów człowieka iszympansa.”
ZOBACZ TEŻ
https://wp-projektu.pl/teksty/teksty-autorskie/podobienstwo-genomow-czlowieka-i-szympansa/
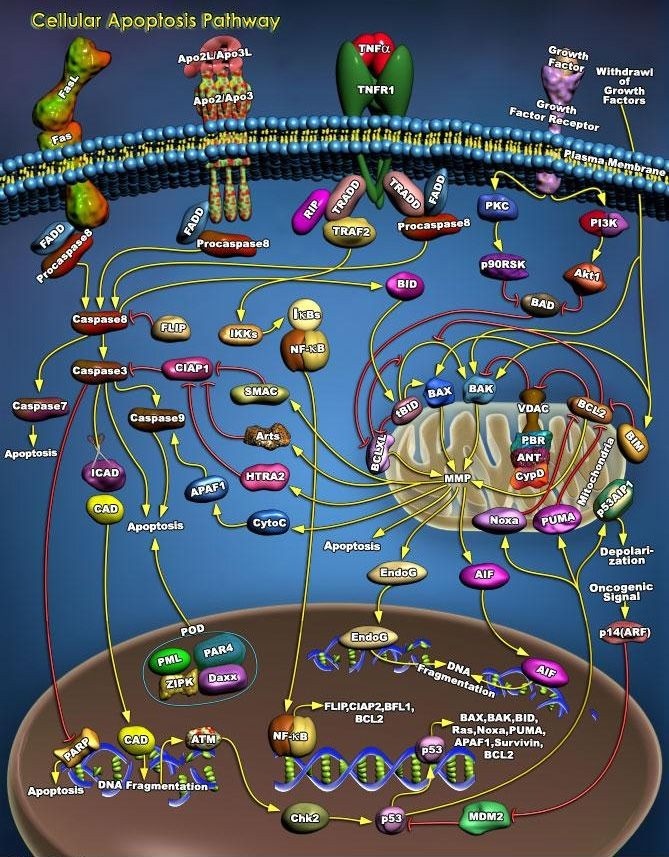
 https://en.wikipedia.org/wiki/Epistasis
https://en.wikipedia.org/wiki/Epistasis
„Epistaza w genomach organizmów występuje w wyniku interakcji między genami. Ta interakcja może być bezpośrednia, jeśli geny kodują białka, które na przykład są oddzielnymi składnikami wieloskładnikowego kompleksu (takie jak rybosom wić bakteryjna, syntaza ATP..), wzajemnie hamują własną aktywność [sprzężenia dodatnie i ujemne], lub jeśli białko kodowane przez jeden gen modyfikuje drugi gen (np. przez fosforylację, albo niezbędną obróbkę potranskrypcyjną, potranslacyjną, modyfikowanie białek za pomocą proteaz - odcinanie sekwencji adresowych, żeby odsłonić sortujące, czy chaperonów ).
Alternatywnie taka interakcja może być pośrednia, w których geny kodują komponenty w szlaku metabolicznym, lub sieci szlaku rozwojowego, szlaku przekazywania sygnałów (transdukcji sygnału), lub kodujące czynniki transkrypcyjne, regulujące ekspresje danego genu w sieci (kolejność, czas wyrażania, co by się stało, gdyby geny kodujące białka rybosomowe nie ulegały precyzyjnej regulacji?). Na przykład gen kodujący enzym, który syntetyzuje penicylinę jest bezużyteczny dla grzyba bez enzymów, które syntetyzują niezbędne prekursory w szlaku metabolicznym.
[…..]
Wysoki poziom epistazy jest zwykle uważany za czynnik ograniczający ewolucję, a ulepszenia wysoce epistatycznej cechy są uważane za mające mniejszą zdolność ewolucji . Dzieje się tak, ponieważ na jakimkolwiek podłożu wielogenowym bardzo niewiele mutacji będzie korzystnych, (!) chociaż może zajść potrzeba wystąpienia wielu mutacji, aby ostatecznie poprawić cechę.”
https://pin.it/7HG7v8xnt
 Pod tymi adresami znajdziesz więcej informacji o oddziaływaniach w ramach szeroko pojętej epistazy i dlaczego jest to problem dla darwinistów:
Pod tymi adresami znajdziesz więcej informacji o oddziaływaniach w ramach szeroko pojętej epistazy i dlaczego jest to problem dla darwinistów:
https://www.salon24.pl/u/slawekp7/1349496,o-pszczolach-higienicznych-i-niehigienicznych
https://www.salon24.pl/u/slawekp7/1333362,istota-hipotezy-doboru-krewniaczego
https://www.salon24.pl/u/slawekp7/1328072,problemy-z-doborem-jednorazowym-i-kumulatywnym-w-genezie-bialek
 https://www.salon24.pl/u/slawekp7/1357970,australopiteki-wymarle-malpy-afrykanskie-nie-byly-przodkami-ludzi
https://www.salon24.pl/u/slawekp7/1357970,australopiteki-wymarle-malpy-afrykanskie-nie-byly-przodkami-ludzi
https://www.salon24.pl/u/slawekp7/1361762,nie-ma-konsensusu-naukowego-odnosnie-przyczyn-wyewoluowania-wlasciwych-ludziom-cech-anatomicznych-i-behawioralnych
 https://kosmos.ptpk.org/index.php/Kosmos/article/view/2433/2364
https://kosmos.ptpk.org/index.php/Kosmos/article/view/2433/2364



O większej ilości problemów z zegarem molekularnym napisano tu:
https://slawekp7.wordpress.com/2018/11/10/glowonogi-edytuja-swoje-rna-hipoteza-zegara-molekularnego-nie-sprawdza-sie-w-przypadku-glowonogow/
https://www.salon24.pl/u/slawekp7/1318613,samolubny-gen-prof-richarda-dawkinsa-czy-samolubne-geny-prof-pawla-golika
 ŹRÓDŁA
ŹRÓDŁA
https://tiny.pl/dq8fq
„O alternatywnym składaniu mówimy wtedy, gdy RNA wytworzone na matrycy określonego genu jest »przycinane« z udziałem enzymów na kilka możliwych sposobów. Można w ten sposób uzyskać kilka różnych produktów (np. białek) na podstawie informacji zakodowanych w pojedynczym genie”.”
https://pubmed.ncbi.nlm.nih.gov/34611352/
„Nowoczesne metody na skalę genomu, które identyfikują nowe geny, takie jak proteogenomika i profilowanie rybosomów, ujawniły, ku zaskoczeniu wielu, że nakładanie się genów, otwartych ramek odczytu, a nawet sekwencji kodujących jest powszechne i funkcjonalnie zintegrowane z genomami prokariotycznymi, eukariotycznymi i wirusowymi . Jednocześnie ograniczenia, jakie nakładające się regiony nakładają na sekwencje genomu i ich ewolucję, można wykorzystać w bioinżynierii do budowy solidniejszych syntetycznych szczepów i konstruktów. Koncentrując się na nakładających się genach kodujących białka i RNA w niniejszym przeglądzie omówiono ich odkrycie, topologię i biogenezę w kontekście biologii genomu. Zwracamy uwagę na nowe i ekscytujące zastosowania nakładania się sekwencji w celu kontrolowania translacji, kompresji syntetycznych konstruktów genetycznych i ochrony przed mutacjami.”
https://tiny.pl/dq8d2
„Czynniki cis to charakterystyczne elementy w sekwencjach intronów oraz eksonów (na granicach z intronami), czynniki trans to różnorodne kompleksy białko - RNA i białka, odpowiedzialne za rozpoznanie intronów, katalizę reakcji składania mRNA oraz jego regulację.”
https://brainly.com/question/27685640
https://quizlet.com/explanations/questions/1-million-proteins-but-human-dna-only-codes-for-25000-1490f817-b7b1-4a6f-929c-60a0b9af9dcb
https://www.statnews.com/2023/11/29/uk-biobank-genome-sequences-500000-people-research/
https://pubmed.ncbi.nlm.nih.gov/11827476/
„Sekwencjonowanie ludzkiego genomu ujawnia, że istnieje około 30 000 genów kodujących jeszcze większą liczbę białek tworzących ludzki proteom.”